Analysis of protein-coding genetic variation in 60,706 humans
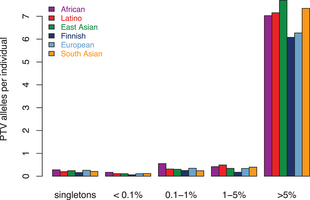
Large-scale reference data sets of human genetic variation are critical for the medical and functional interpretation of DNA sequence changes. Here we describe the aggregation and analysis of high-quality exome (protein-coding region) DNA sequence data for 60,706 individuals of diverse ancestries generated as part of the Exome Aggregation Consortium (ExAC). This catalogue of human genetic diversity contains an average of one variant every eight bases of the exome, and provides direct evidence for the presence of widespread mutational recurrence. We have used this catalogue to calculate objective metrics of pathogenicity for sequence variants, and to identify genes subject to strong selection against various classes of mutation; identifying 3,230 genes with near-complete depletion of predicted protein-truncating variants, with 72% of these genes having no currently established human disease phenotype. Finally, we demonstrate that these data can be used for the efficient filtering of candidate disease-causing variants, and for the discovery of human ‘knockout’ variants in protein-coding genes.